
Currently, there is no cure for Parkinson's disease. While several treatment options are available to manage symptoms, targeted molecular therapies are still in development. Our laboratory focuses on understanding the molecular basis of Parkinson's disease.
BACKGROUND
The genetic component of Parkinson’s disease
Parkinson's disease (PD) was traditionally considered idiopathic, but recent research highlights its genetic components. The first mutations identified in PD were in the α-synuclein gene (SNCA), which is linked to the formation of Lewy bodies, a key pathological feature of the disease.
In 2004, mutations in the LRRK2 gene gained attention due to their prevalence in patients with PD worldwide. LRRK2-related PD typically presents similarly to common non-genetic PD and is typically late-onset, in contrast to other genes linked to early-onset forms.
Genome-wide association studies have shown that genes such as SNCA and LRRK2, which are associated with inherited PD, can also act as risk factors for sporadic PD. Understanding how these mutations affect cellular processes can elucidate the mechanisms underlying sporadic PD.
WHAT WE ARE INTERESTED IN
Investigating the cell type-specific effects of PD-associated mutations on neuronal synapses and the circuits they form.
OUR APPROACH
We aim to identify early and reversible dysfunctions before terminal pathology resulting from mutations in specific cellular subtypes, thereby enhancing our understanding of targetable dysfunctions related to PD.
We utilize advanced genetic mouse models, combined with a range of techniques, including super-resolution imaging, refined proteomics and biochemical methods, translatomics, electrophysiology, anatomical methods, and behavioral assays.
MAIN Findings
LRRK2 and α-synuclein act synergistically in the molecular and cellular events that contribute to PD. (Lin*, Parisiadou* et al., Neuron, 2009)
LRRK2 directs neuronal cytoskeletal dynamics— implications for synapse structure and function. (Parisiadou et al., Journal of Neuroscience, 2009; Parisiadou et al., Nature Neuroscience, 2014)
Different effects of LRRK2 mutations on excitatory synapses in various subtypes of striatal neurons. (Chen…Parisiadou, eLife, 2022; Xenias…Parisiadou, Communications Biology, 2022, Chen… Parisiadou, Molecular Psychiatry, 2025)
LRRK2 regulates the in vivo dopamine signaling of the most vulnerable dopamine neuron subpopulation in PD. (Chen…Parisiadou, BiorXiv, 2025)

Dopamine release sites of identified dopamine neuron subtypes with varying susceptibility in PD. 3D super-resolution imaging demonstrates the simultaneous labeling of axons from two distinct dopamine neuron subpopulations in the striatum. The bassoon protein, a marker for dopamine release sites, is illustrated within these neurons, as shown by Chen…Parisiadou, BiorXiv, 2025

Anatomical features of the synapse of genetically labeled striatal neurons. Example of confocal maximum projection image of an Adora2aCre (indirect pathway) SPN expressing AAV5/DIO-EGFP. Right, Examples of dendritic segments in different treatment conditions. From Chen…Parisiadou. Molecular Psychiatry, 2025

Physiological properties of identified neurons in the striatum: a synapse-specific analysis (A) A schematic of the experimental design for measuring glutamate uncaging-evoked currents in spiny projection neurons (SPNs) of the striatum. (B) An example projection from a two-photon microscopy stack shows a GFP+ SPN from the indirect pathway. (C)Close-up images of SPN dendrites across different genotypes and subtypes are displayed using an inverse greyscale LUT. The RC and GS indicate pathogenic LRRK2 mutations R1441C and G2019S. From Chen…Parisiadou, eLife, 2020

Measurement of dopamine release in mice with LRRK2 pathogenic mutations. Representative time courses for dopamine [DA] with corresponding cyclic voltammograms and color maps of fast-scanning cyclic voltammetry recordings in the striatum. Colormaps are normalized to the oxidation current from the WT. RC, GS; R1441C and G2019S pathogenic mutations. Xenias…Parisiadou, Communications Biology, 2022.
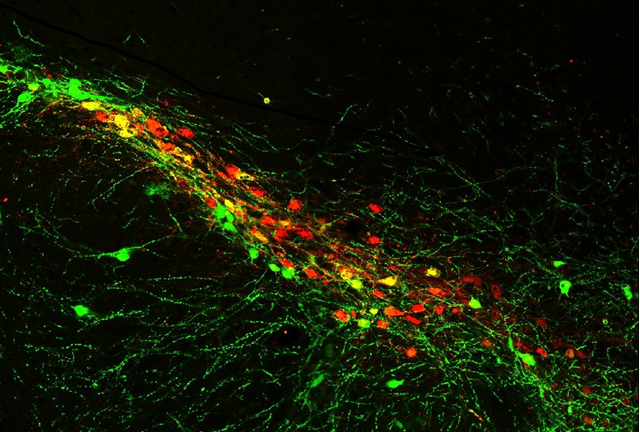
Mouse brain section displaying dopamine neurons in the substantia nigra pars compacta. These dopamine neurons are particularly vulnerable in Parkinson’s disease.
WHY DO WE STUDY LRRK2?
Provided the importance of LRRK2 in both familial and sporadic PD, modulation of its dysfunction in the disease context would be a way to intervene in PD more broadly. The LRRK2 protein consists of a GTPase domain known as the Ras of complex (ROC) domain, along with a kinase domain (see Figure 1). This unique combination makes LRRK2 a promising target for drug development. Currently, LRRK2 is the focus of ongoing clinical trials to develop disease-modifying therapies for Parkinson's disease. Consequently, our primary scientific efforts focus on understanding both the physiological role of LRRK2 and its dysfunction in disease.
To achieve these research goals, the laboratory employs a multidisciplinary approach spanning cellular, network, and behavioral levels across several model systems.

Figure 1: LRRK2 domains and pathogenic mutations. LRRK2 is a large and multi-domain protein. Its central enzymatic region consists of a GTPase Ras of complex (ROC domain), a C-terminal of Roc domain (COR), and a kinase domain. Different protein-protein interaction domains surround the enzymatic core. The pathogenic mutations that definitively cause PD are restricted to the central catalytic region of LRRK2.
Our core scientific values include sharing unpublished data early, promoting collaboration, and depositing datasets in publicly accessible repositories to ensure transparency.
While much remains to be learned about Parkinson's disease, research into α-synuclein and LRRK2 is helping to elucidate its complex origins.
These findings not only enhance our understanding of the disease but also open the door to innovative therapies that target the underlying mechanisms rather than just managing symptoms.